
Die Europäische Kommission hat 2017 zwei neue Verordnungen erlassen: zum einen die Medizinprodukteverordnung 2017/745/EU, kurz MDR. Und zum anderen die In-vitro-Diagnostika-Verordnung 2017/746/EU, genannt IVDR. In den letzten 20 Jahren konnten in beiden Bereichen viele Fortschritte erzielt werden. Höchste Zeit also, die erreichten Ziele zu manifestieren, Schlüsselelemente anzupassen und die Inhalte für alle transparenter zu machen – vom Hersteller bis zum Patienten.
Zukünftig wird die MDR die bestehende Medizinprodukterichtlinie (93/42/EWG – MDD) und die Richtlinie über aktive implantierbare Medizinprodukte (90/385/EWG – AIMDD) ersetzen. Wichtig zu wissen: Im Gegensatz zu Richtlinien müssen europäische Verordnungen nicht auf das jeweilige nationale Recht übertragen werden. Das bedeutet, dass die Umsetzung der Verordnungen für alle EU-Mitgliedstaaten gleichermaßen erfolgt.
Veröffentlicht wurde MDR schon im Mai 2017. Damit fiel der Startschuss für eine dreijährige Übergangsphase, in der AIMDD und MDD von der neuen Medizinprodukteverordnung abgelöst werden sollen. Gültig ist die MDR ab Mai 2020, ab diesem Zeitpunkt können Akteure bereits freiwillig nach der Verordnung vorgehen. Bis November 2021 müssen dann die Daten gemäß der neuen MDR hochgeladen worden sein. Während die MDR bereits ab Mai 2020 gültig sein wird, tritt die IVDR erst 2022 in Kraft. Jetzt gilt es für alle Akteure, die an Medizinprodukten beteiligt sind, sich nach beiden Verordnungen richten.
Was wird sich für Sie ändern?
Wenn aus einer Richtlinie eine Verordnung wird, spricht man eher von neuen Anforderungen, als von Änderungen. Denn die bereits eingeschlagene Richtung bleibt bestehen. Das bedeutet im Fall der neuen Medizinprodukteverordnung: Die bisherigen Anforderungen bleiben alle bestehen, dafür sind einige Neuerungen hinzugekommen. Hier ein Auszug der wichtigsten Punkte:
- Einführung eines neuen, einzigartigen Identifizierungssystems (UDI – Unique Device Identification)
- Benennung einer Person, die für die Einhaltung der Vorschriften verantwortlich ist.
- Benennung eines Bevollmächtigten, wenn der Hersteller seinen Sitz außerhalb der EU hat (Authorized Representative)
Wo setzt EUDAMED ganz genau an?
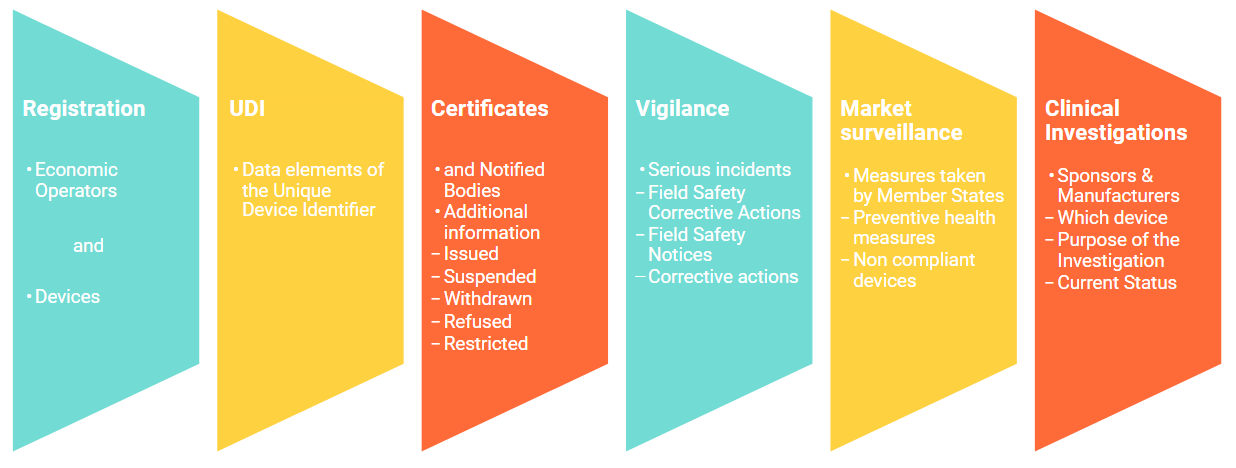
Fest steht: Die Identifizierung von zugelassenen Geräten ist eins der Hauptelemente, die aus den neuen Verordnungen hervorgeht. Das bringt natürlich hohe Datenmengen mit sich. Um die Quantität und auch die Qualität der gesammelten Daten sicher und optimal managen zu können, wurde eine Europäische Datenbank für Medizinprodukte entwickelt, „EUDAMED“. Bei dieser European Database on Medical Devices handelt es sich um ein Internetportal, das die Hersteller für die Eingabe und Pflege ihrer Medizinprodukte eigenverantwortlich nutzen. Diese Datenbank gliedert sich in folgende Module:
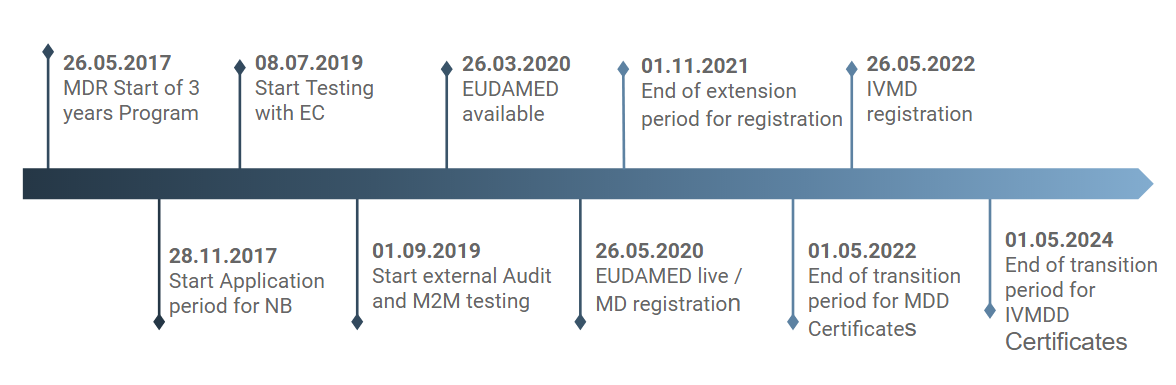
Welche wichtigen Termine sollten Sie notiert haben?
Damit Sie einen genauen Überblick über die nächsten Meilensteine erhalten, finden Sie hier die wichtigsten Termine und Fakten (Stand August 2019).
Wo stehen wir jetzt?
Seit Mai 2017 ist viel passiert. Darum können wir zu einigen Punkten bereits konkrete Aussagen treffen:
- Für die Produktregistrierung wurde eine Verlängerung um 18 Monate gewährt. Allerdings gibt es hier einige Ausnahmen, die hauptsächlich die klinische Überwachung und die Patientensicherheit betreffen (bis 11/2021).
- EUDAMED wird bei der Aktivierung nicht voll funktionsfähig sein. Anhand einer Roadmap können die Beteiligten das Timing für die Implementierung exakt nachvollziehen, geplant sind hier 4 Releases:
– High (1): erstes Release im März 2020
– High (2): zweites Release im November 2020
– Medium (3): drittes Release im Mai 2021
– Medium (4): Viertes Release im Mai 2022
– Low (5): späteres Release (not for audit)
Und atrify?
- atrify nimmt aktiv am Austausch zwischen Herstellern, der Europäischen Kommission und MedTech Europe teil. MedTech vertritt dabei die Medizintechnikbranche.
- atrify arbeitet an der AS4-Verbindung und bereitet aktuell die XML-Datei vor. Diese Datei wird als Grundlage für die Kommunikation mit EUDAMED dienen.
Es ist für uns sehr spannend zu erleben, wie die Implementierung voranschreitet. Wir wissen, wie entscheidend die Umsetzung für alle Beteiligten ist und freuen uns auf die Realisation der nächsten Schritte. Natürlich stellt die Einhaltung der MDR die Hersteller vor eine neue Herausforderung. Darum setzen wir alles daran, die Hersteller mit unseren Lösungen gezielt zu unterstützen.
Wie geht es weiter?
Denn wie Sie wissen, stehen wir erst am Anfang der EUDAMED-Geschichte. Deshalb halten wir Sie zukünftig auf dem Laufenden und informieren Sie regelmäßig über Änderungen, Neuigkeiten und Verbesserungen.
Erfahren Sie mehr über unsere EUDAMED Lösung.



